
More Information
Submitted: 06 October 2019 | Approved: 19 October 2020 | Published: 20 October 2020
How to cite this article: Nabavi M, Rezaeifar P, Fallahpour M, Arshi S, Bemanian MH, et al. Cystic fibrosis and congenital adrenal hyperplasia: A rare occurrence with diagnostic dilemmas, similarities and contradictions. Arch Asthma Allergy Immunol. 2020; 4: 018-020.
DOI: 10.29328/journal.aaai.1001021
ORCID: orcid.org/0000-0002-1435-2811
Copyright License: © 2020 Nabavi M, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Cystic fibrosis; Congenital adrenal hyperplasia; Sweat test

Cystic fibrosis and congenital adrenal hyperplasia: A rare occurrence with diagnostic dilemmas, similarities and contradictions
Mohammad Nabavi1, Parisa Rezaeifar2, Morteza Fallahpour1, Saba Arshi2, Mohammad Hassan Bemanian1, Sima Shokri1 and Afshin Rezaeifar1*
1Department of Allergy and Clinical Immunology, Iran University of Medical Sciences, Tehran, Iran
2TB and Lung Disease Research Center, Tabriz University of Medical Sciences, Tabriz, Iran
*Address for Correspondence: Afshin Rezaeifar, Department of Allergy and Clinical Immunology, Iran University of Medical Sciences, Tehran, Iran, Tel&Fox: +982166554933, Email: [email protected]
Cystic fibrosis (CF) is a hereditary syndrome composed of exocrine gland dysfunction involving multiple systems which if untreated may result in chronic respiratory infections, pancreatic enzyme deficiency and failure to thrive. The association between CF and other inherited diseases or congenital anomalies is rare. We describe a rare case of CF with concomitant congenital adrenal hyperplasia (CAH). 21- Hydroxylase deficiency accounts for most CAH cases. Varity in clinical phenotypes depends on the amount of enzymatic activity which in turn depends on different combination of gene mutations. The genes of CAH and CF are located in different locations. The chance of these diseases coexisting in our patient would be a rare combination. However, such a case will be more frequent in our population than others because of consanguineous marriage and common ancestors. There are diagnostic difficulties, similarities and contradictions between two diseases and they are pointed out.
Cystic Fibrosis (CF) is an autosomal recessive disease believed to arise from abnormal mucus plugging in exocrine ducts. CF gene is located on chromosome 7 and encodes a cAMP-regulated chloride channel called CF Transmembrane Conductance Regulator (CFTR). CFTR is expressed in many epithelial cells including sweat ducts, airways, pancreatic ducts, intestine, biliary tree and vas deference. CFTR dysfunction results in ionic imbalance of epithelial secretions in several organs and related clinical manifestations [1-3]. The prevalence of CF varies considerably among different populations around the world, with an incidence of about 1 in every 2500 persons. There are limited studies regarding the prevalence of CF in Iran [4].
Congenital adrenal hyperplasia (CAH) is another autosomal recessive disease. 21- Hydroxylase deficiency accounts for 90% - 95% of CAH cases. Based on the phenotypic expression, it is categorized into two major forms: severe or classic form, and late onset or non-classic form. The former, consists of salt wasting or simple virilizing type. Varity in clinical phenotypes depends on reduced enzymatic activity due to different combination of gene mutations which is located on chromosome 6 (locus 6p21.3) [5,6]. The incidence of 21-Hydroxylase deficiency is 1: 15000 to 1: 10000 live births. Prevalence of CAH tends to be high in some countries like Iran with frequent consanguineous marriages [5].
Simultaneous occurrence of CF and other inherited diseases or congenital anomalies has been seldom described [7]. These associations comprising syringomyelia [8], lymphoblastic leukemia [9], gastrointestinal cancer [10], intra-sylvian fibroma [11], congenital generalized hamartoma [12], infantile hypertrophic pyloric stenosis [13], sickle cell disease [7,14], CAH [7,15], Ehlers Danlos syndrome [7,16,17] and Beckwith-Wiedemann syndrome (BWS) [18].
This manuscript is a report of an 8-year-old boy with rare concurrence of CF and CAH and a brief literature review.
An 8-year-old boy with CAH is referred to an allergist due to history of chronic cough, refractory rhinitis and frequent pulmonary infections to assess immunodeficiency disorders. In medical history, CAH was suspected in 18-day-old due to skin hyperpigmentation and electrolytes imbalance. Genetic analysis confirmed CAH with 21-Hydroxylase deficiency. Chromosomal study revealed normal male karyotype with 46XY. In family history we noticed that he is the result of consanguineous marriage. He used to consume hydrocortisone and fludrocortisone tablets for CAH.
In review of systems there was no history of vomiting, diarrhea or hyperhidrosis but multiple hospitalizations due to pneumonia. On examination, we detected wheeze and crackle in lung auscultation, post nasal discharge in oropharynx and clubbing in fingers, but no skin hyperpigmentation or ambiguous genitalia.
In laboratory studies, normal amounts of serum electrolytes was detected and chloride tests were in borderline and normal ranges (53 mmol/lit and 36 mmol/lit). Stool test for fat drop was negative. Blood gases analysis and O2 saturation were normal. There was no problem in Immunological workup including immunoglobulins levels, IgG subclasses, antibody responses to vaccines and flow cytometery. Saccharin blue test was done to evaluation of cilliary dyskinesia and the result was normal. In imaging studies, computed tomography (CT) scan revealed normal chest but congestion in maxillary and ethmoidal sinuses accompany with bone attenuation and remodeling in paranasal sinuses (Figure 1A,B). Due to strong suspicion of CF, CFTR study was carried out and was found to have C 3717+ 12191C>T homozygous that is a class V mutation.
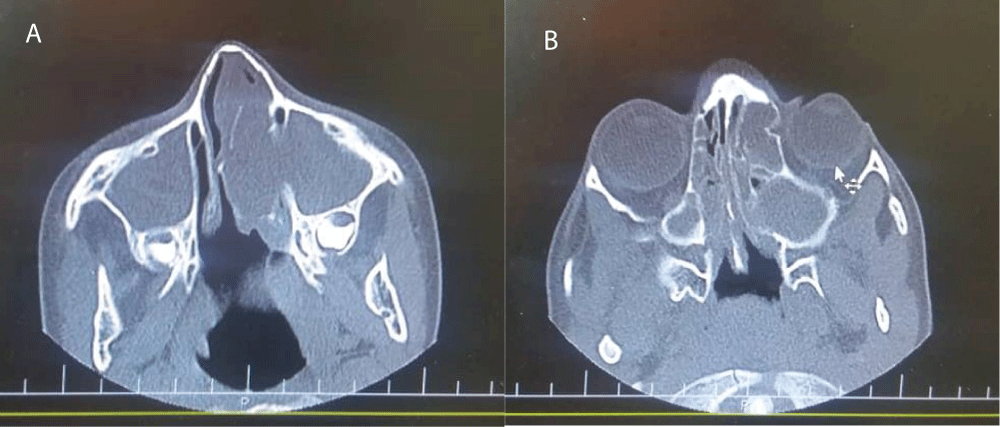
Figure 1: A, B: Paranasal sinuses computed tomography scan show congestion in maxillary and ethmoidal sinuses accompany with bone attenuation and remodeling in paranasal sinuses.
Diagnosis of CF in our patient was made after careful review of his history, clinical presentations and genetic analysis. Pancreatic enzymes, fat soluble vitamins, prophylactic antibiotics, inhaled corticosteroid and bronchodilator and inhaled weekly Amikacin were added to his drug list. His clinical condition was improved progressively. Meanwhile, the patient is 9-year-old and due to CF and CAH is under care and control.
CF and CAH are diseases with autosomal recessive inheritance. As far as we know, combination of CF and CAH has previously reported in Switzerland and Saudi Arabia studies [7,15].
Both diseases may be the same of the delayed physical development. Both of them present with FTT, poor feeding, vomiting and dehydration. Pulmonary manifestations are major complications in CF; also CAH may have affected the progression of disease due to recurrent vomiting and aspiration with progressive lung disease [1].
Cystic fibrosis itself tends to electrolyte abnormalities like CAH, such as hypochloremia, hyponatremia, hypokalemia or hyperbicarbonatemia that are almost always associated with often clinically in apparent fluid volume depletion. Excessive salt loss in CF predisposes to metabolic alkalosis and heat prostration [2].
Medical records of all confirmed cases with CF are based on high sweat chloride test or identification of CFTR gene mutation in genetic analysis. The false positive or false negative sweat test results of cases were reported [1,3]. The results of sweat test of our patient were found to be among normal or borderline range the test was repeated twice. The CFTR genotyping analysis by method of PCR with reverse hybridization was detected C 3717+ 12191C>T homozygous and diagnosis was confirmed with the clinical findings. We speculate that these mutations may lead to negative results on sweat test. C 3717 + 12191C>T is a mutation in intron 22. Legacy name is 3849+10KbC>T. This mutation at first was found in a female with mild CF and normal values in sweat tests in a Pakistani family with consanguineous mating. This mutation is a class V mutation which normal CFTR protein is created and moved to the cell surface but in insufficient quantities. Classes IV, V mutations cause a reduction in function and have a milder effect in opposition to classes I, II, and III mutations that generally lead to complete loss of function and a more severe disease [19]. The causes of the false negative results on sweat test are indicated as technical reasons, specific mutations, hypohydrotic ectodermal dysplasia, inadequate sweat collection, mineralocorticoid treatment, young age, edema, hypoproteinemia and penicillin treatment [1]. Mineralocorticoid treatment due to CAH and young age are the most common reasons and this specific mutation may be responsible for our negative sweat test result. Up to date, based on genotype-phenotype correlations, we are aware that some CFTR mutations can cause unusual electrophysiological or clinical manifestations. In conclusion although the sweat test is the most commonly used method for the diagnosis of CF, it does not always give a clear answer. Mutation analysis may be helpful when clinical findings are suggestive of cystic fibrosis, even if sweat test results are negative [1].
Diagnose of CAH is based on physical exams, measurement of hormone levels produced by the adrenal gland-cortisol, aldosterone and androgen in blood and karyotyping [5,6]. In classic 21-hydroxylase deficiency, laboratory studies will show hypoglycemia, hyponatremia, hyperkalemia, metabolic acidosis and can also be confirmed biochemically by a very high concentration of 17-hydroxyprogesterone. False-positive results are common with premature infants. False negative results may occur if samples are drawn late in the afternoon as adrenal hormones exhibit diurnal variation. The gold standard for hormonal diagnosis is the corticotropin stimulation test. Karyotyping is a blood analysis that can be done to analyze chromosomes to identify genetic sex. Genetic analysis can be helpful to confirm a diagnosis of CAH but it is not necessary if classic clinical and laboratory findings are present [5,6].
The gene of CAH (21-hydroxylase deficiency) and CF must be taken to have different locations. The former gene is on chromosome 6 that is closely linked to the HLA region and the latter one is located on chromosome 7 whereas similar linkage to the HLA region is very unlikely. However, there is a little likelihood of linked inheritance under normal situations. Possibility between the interactions of the genes at the two loci is to be considered of an independent “inborn error of metabolism” originating in an unknown genotype of one of the two loci [1,7,20].
The chance of these diseases coexisting in our patient would be in the range of 1/2500 * 1/10000 = 1/25000000 live births, i.e. it will be a rare combination. This calculation assumes both diseases are inherited in an autosomal recessive pattern in which both parents contribute on abnormal allele for CAH and CF to the child. However, such a case will be more frequent in our population than others because of consanguineous marriage and common ancestors [7,20].
We appreciate the patient’s parents for participating in this study and Miss Nadieh Akbarpour in Department of Allergy and Clinical Immunology, Iran University of Medical Sciences for helping us.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Ethical considerations and consent to participate
All ethical principles were considered in this article. The parents of patient enrolled in the study gave their written informed consent.
- Smyth AR, Bell SC, Bojcin S, Bryon M, Duff A, et al. European cystic fibrosis society standards of care: best practice guidelines. J Cystic Fibrosis. 2014; 13: S23-42. PubMed: https://pubmed.ncbi.nlm.nih.gov/24856775/
- Davis PB. Cystic fibrosis since 1938. Am J Respiratory Critical Care Med. 2006; 173: 475-482. PubMed: https://pubmed.ncbi.nlm.nih.gov/16126935/
- Derichs N. Targeting a genetic defect: cystic fibrosis transmembrane conductance regulator modulators in cystic fibrosis. European Respiratory Review. 2013; 22: 58-65. PubMed: https://pubmed.ncbi.nlm.nih.gov/23457166/
- Aghamohammadi A, Keivanfar M, Navaei S, Shirzadi R, Masiha F, et al. First cystic fibrosis patient registry annual data report-cystic fibrosis foundation of Iran. Acta Medica Iranica. 2019; 57: 33-41.
- Rabbani B, Mahdieh N, Ashtiani MT, Akbari MT, Rabbani A. Molecular diagnosis of congenital adrenal hyperplasia in Iran: focusing on CYP21A2 gene. Iranian J Pediatrics. 2011; 21: 139-150. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3446151/
- Childs B, Grumbach MM, Van Wyk JJ. Virilizing adrenal hyperplasia; a genetic and hormonal study. J Clini Investig. 1956; 35: 213-222. PubMed: https://pubmed.ncbi.nlm.nih.gov/13286340/
- Banjar HH. Cystic fibrosis: presentation with other diseases, the experience in Saudi Arabia. J Cystic Fibrosis. 2003; 2: 155-159. PubMed: https://pubmed.ncbi.nlm.nih.gov/15463866/
- Rusakow LS, Guarín M, Lyon RM, Splaingard ML. Syringomyelia and chiari malformation presenting as scoliosis in cystic fibrosis. Pediatric Pulmonol. 1995; 19: 317-318. PubMed: https://pubmed.ncbi.nlm.nih.gov/7567208/
- Rizzari C, Conter V, Jankovic M, D'Angelo P, Masera G, et al. Acute lymphoblastic leukaemia in a child with cystic fibrosis. Haematologica. 1992; 77: 427-429. PubMed: https://pubmed.ncbi.nlm.nih.gov/1483594/
- Schöni MH, Maisonneuve P, Schöni-Affolter F, Lowenfels AB. Cancer risk in patients with cystic fibrosis: The European data. CF/CSG Group. J Royal Society Med. 1996; 89(Suppl 27): 38. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1295624/
- Pollack IF, Hamilton RL, Fitz C, Orenstein DM. An intrasylvian “fibroma” in a child with cystic fibrosis: case report. Neurosurgery. 2000; 46: 744-748.
- Mascaró JM, Ferrando J, Bombi JA, Lambruschini N. Congenital generalized follicular hamartoma associated with alopecia and cystic fibrosis in three siblings. Arc Dermatol. 1995; 131: 454-458. PubMed: https://pubmed.ncbi.nlm.nih.gov/7726589/
- Kakish KS. Cystic fibrosis and infantile hypertrophic pyloric stenosis: Is there an association? Pediatric pulmonology. 2002; 33: 404-405.
- Chang J, Kan Y. Antenatal diagnosis of sickle cell anaemia by direct analysis of the sickle mutation. The Lancet. 1981; 318: 1127-1129. PubMed: https://pubmed.ncbi.nlm.nih.gov/6118575/
- Nielsen Oh, Haahr J. Adrenogenital Syndrome and Cystic Fibrosis. Acta Pædiatrica. 1982; 71: 339-341. PubMed: https://pubmed.ncbi.nlm.nih.gov/7136645/
- Dowton SB, Pincott S, Demmer L. Respiratory complications of Ehlers‐Danlos syndrome type IV. Clin Genet. 1996; 50: 510-514. PubMed: https://pubmed.ncbi.nlm.nih.gov/9147885/
- Jarisch A, Giunta C, Zielen S, König R, Steinmann B. Sibs affected with both Ehlers‐Danlos syndrome type VI and cystic fibrosis. Am J Med Genet. 1998; 78: 455-460. PubMed: https://pubmed.ncbi.nlm.nih.gov/9714013/
- Aguiar C, Correia-Costa L, Eden P, Guedes-Vaz L. Cystic fibrosis and beckwith-wiedemann syndrome: A case report. J Clin Med Res. 2015; 7: 186-188. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4285066/
- Ivanov M, Matsvay A, Glazova O, Krasovskiy S, Usacheva M, et al. Targeted sequencing reveals complex, phenotype-correlated genotypes in cystic fibrosis. BMC Med Genom. 2018; 11: 13. PubMed: https://pubmed.ncbi.nlm.nih.gov/29504914/
- Lamm LU, Thorsen IL, Petersen GB, Jorgensen J, Henningsen K, et al. Data on the HL-A linkage group. Ann Human Genetics. 1975; 38: 383-.390.